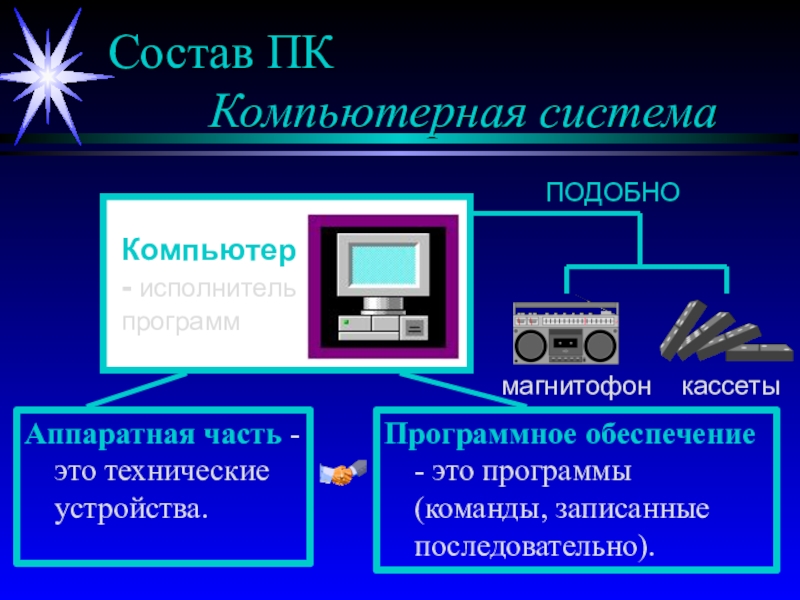
фазовых превращений,
направление протекания процессов в физико-химических системах,
химические и
фазовые равновесияВозможность протекания процесса – термодинамика
2Н2 + О2 = 2Н2О
По т/д расчетам – возможна при любых Т (ниже 5000К) и давлении, близком к атмосферному
В реальности смесь газов не взаимодействует, пока нет Кт (Pt)
Скорость протекания процесса - кинетика












 – энтальпия образования CO (г)ΔН(2) – энтальпия образования CO2 (г)Следствие")

 и энтальпийная диаграмма (б) для изменения энтальпии реакции газообразного")

-энтальпия образования данного вещества ABn из атомовABn =")


")
 сгорания (combustion)")

, исходя из ΔfН0(298) = 0, найдено ΔhН0(298) = -1075")



(например, когда неизвестна теплота образования одного из реагентов)Результат")


 и продуктов (2)Пример")



растет при переходе тв – ж – гв аморфном состоянии выше,")





 предложил определить это")
- характеристика устойчивости системы при постоянном")

 < 0Для самопроизвольного протекания процесса при")







К.р. сходно с Истинным р. по")


 влияют не на величину константы равновесия,")


:Изменения ΔG в реальных условиях химических")



гетерогенная (на поверхности раздела фаз,")



![Для реакции разложения Н2О2:v = k[Н2О2]1Кинетические уравнения для реакций с различным порядком2Н2О2 = 2Н2О + О2и убедились,](/img/thumbs/59493acb0217b1164c35ed0cd2aaead4-800x.jpg "Начала химической термодинамики
Х.Т. – раздел химии, изучающий
энергетику Для реакции разложения Н2О2:v = k[Н2О2]1Кинетические уравнения для реакций с различным")

![Физический смысл величины k:константа скорости реакции численно равна скорости реакции при единичных концентрациях реагентовv = k[C]m[AB]n](/img/thumbs/a70d757d8351772b81a02bb7e0c32452-800x.jpg "Начала химической термодинамики
Х.Т. – раздел химии, изучающий
энергетику Физический смысл величины k:константа скорости реакции численно равна скорости реакции при")

")
![«Кинетический» вывод константы равновесияДля обратимой одностадийной реакции А + В АВvпр = k[А][B]можно записать выражениезатем проявится](/img/tmb/3/261711/cb972b300114cde7f8d2d45398157e0c-800x.jpg "Начала химической термодинамики
Х.Т. – раздел химии, изучающий
энергетику «Кинетический» вывод константы равновесияДля обратимой одностадийной реакции А + В ")


















лимитирующая стадияH2O22 OH*OH* + HIПример:I")
А+ВMNА+Cикогда одна из реакций индуцируется другойПример:HI + H2CrO46")